


Glucose and other nutrients regulate many aspects of pancreatic islet physiology. This includes not only insulin release, but also insulin synthesis and storage and other aspects of β-cell biology, including cell proliferation, apoptosis, differentiation, and gene expression. This implies that in addition to the well-described signals for insulin release, other intracellular signaling mechanisms are needed. Here we describe the role of global and local Ca2+ signals in insulin release, the regulation of these signals by new membrane receptors, and the generation of nuclear Ca2+ signals involved in gene expression. An integrated view of these pathways should improve the present description of the β-cell biology and provide new targets for novel drugs.
Pancreatic β-cell physiology is mostly conducted by glucose and other nutrients. Indeed, glucose regulates not only insulin release, but also insulin synthesis and storage, cell proliferation, apoptosis, differentiation, and gene expression. A physiological analysis of the effects of glucose, and other nutrients, should consider these different cell processes. Therefore, the proper physiological response of the cell requires a rigorous control of all these variables. Accordingly, various biochemical cascades acting in a concerted manner establish a complex system of information delivery in which cell messages are coordinated precisely to activate cell processes in a specific mode. Recent findings have opened original perspectives in pancreatic β-cell signal transduction. Those include the existence of receptors for novel extracellular messengers and the new location of ion channels. Here, we aim to review these new events and to integrate them within the global scenario of β-cell signaling.
IMPORTANCE OF LOCAL Ca2+ SIGNALS IN REGULATED INSULIN SECRETION
Changes in the intracellular Ca2+ concentration ([Ca2+]i) in response to different stimuli play a pivotal role in signaling numerous cellular processes. The versatility of Ca2+ to relay cellular information is achieved by different ways that manage intracellular Ca2+ signals, which include location and/or spatial constraint, amplitude, frequency, and duration (1). In β-cells, as in the majority of cells, Ca2+ signals play a key role as a messenger for multiple functions. The classic stimulus-secretion coupling that drives insulin release involves the closure of plasma membrane ATP-dependent K+ (KATP) channels by increasing the intracellular ATP/ADP ratio (2), diadenosine polyphosphates (DPs) (3,4), and cGMP (5) on account of glucose metabolism (Fig. 1). Channel closure induces membrane depolarization that activates voltage-operated Ca2+ (VOC) channels and Ca2+ influx (6). The set of channels of β-cell plasma membrane generates an oscillatory electrical activity that causes [Ca2+]i to oscillate (7–9). Remarkably, the oscillatory Ca2+ pattern triggers a pulsatile insulin secretion (10–12). Membrane potential and [Ca2+]i oscillations are built as a result of the precise interplay between gap-junction conductance and cell input conductance in the appropriate range (13,14).
The stimulus-secretion coupling is regulated not only by global Ca2+ oscillations, as described in the above paragraph, but by localized Ca2+ signals as well. These occur beneath the plasma membrane in areas associated with hot spots of exocytosis (15–17). There are several advantages of developing spatial-restricted Ca2+ signals rather than global increases in Ca2+ to control insulin release. Localized Ca2+ signals have only a limited spatial spread, and [Ca2+]i declines sharply away from the site of origin (i.e., VOC channels) because of the buffering properties of the cytosol. This results in high [Ca2+]i near the plasma membrane Ca2+ channels and adjacent Ca2+-sensitive targets in the exocytotic machinery. As a consequence, the Ca2+ signal has a highly specific and efficient effect on secretion, probably by ensuring the precise [Ca2+]i required for the activation of the low-affinity Ca2+-dependent exocytotic enzymes (17,18) and preventing a massive activation of other Ca2+-dependent functions distant from the sites of exocytosis. In the pancreatic β-cell, as in other cell types, the secretory granules exist in distinct functional pools, with different proximity to the plasma membrane and probably with different exposure to [Ca2+]i (19). One of them is the readily releasable pool (RRP). A group of these RRP granules is located in close proximity to VOC channels, and, consequently, they should be exposed to exocytotic levels of Ca2+ during the opening of Ca2+ channels. These granules are referred to as the immediate releasable pool and are situated ∼10 nm from Ca2+ channels. At this distance, the Ca2+ concentration can be expected to rise to high levels, probably in the micromolar range (15,16), required by proteins participating in membrane fusion and exocytosis (17,18). Both RRP and immediate releasable pool granules interact with the plasma membrane via exocytotic proteins, which mediate granule release and thus insulin secretion (20,21). Therefore, because release-competent granules are situated close to the points of Ca2+ entry, the development of localized submembranal Ca2+ gradients would dramatically increase the efficiency and specificity of Ca2+ to trigger the release of these granules. Another advantage of these local Ca2+ transients is that they can be removed from the cytoplasm at relatively low energy cost for the cells, in contrast to global Ca2+ changes. This is particularly relevant in the pancreatic β-cell because Ca2+ removal from the cytoplasm is mostly driven by Ca2+ ATPases. Consequently, ATP consumption would produce a fall in the ATP/ADP ratio, which could affect the KATP channel activity and thereby the membrane potential. Finally, because secretion in the pancreatic β-cell can be maintained over periods of several minutes, the spatial restriction of Ca2+ signals close to the plasma membrane for insulin release would prevent a broad activation of other Ca2+-dependent functions. In fact, prolonged high cytosolic Ca2+ levels may trigger other cellular processes including apoptosis, which would lead to cell death.
How were submembranal local Ca2+ signals determined in β-cells? In the early 1990s, two important findings suggested the existence of localized Ca2+ gradients. On the one hand, agonists such as carbachol were not able to initiate insulin release when applied in the absence of glucose, even when they induced higher global [Ca2+]i signals than glucose (22). On the other hand, there were discrepancies between the Ca2+ sensitivity of exocytotic responses and the levels of [Ca2+]i reached in the cytosol during glucose stimulation (22–25). Therefore, it was suggested that high [Ca2+] changes restricted to discrete domains beneath the cell membrane could have principal control on the secretory machinery and thus could be responsible for triggering exocytosis of insulin granules. In light of these new ideas, novel digital imaging methods that drastically increase the image spatial resolution by removing the out-of-focus fluorescence, have allowed us to measure the development of submembranal [Ca2+]i gradients in pancreatic islet cells in response to glucose and carbachol (22). That study showed that 1) stimulatory glucose concentrations induced steep spatial gradients of [Ca2+]i in the vicinity of the plasma membrane, 2) Ca2+ levels in response to glucose reached values in the micromolar range, 3) these local Ca2+ signals were polarized in the cell, and 4) carbachol induced [Ca2+]i changes that originated in the center of the cell and declined toward the cell membrane. These results indicated that insulin release might be principally regulated by [Ca2+]i in the vicinity of the exocytotic sites. New evidence has been accumulated in favor of this idea. Actually, in the pancreatic β-cell, there is a faint coupling of Ca2+ channels to the secretory machinery, with exocytosis being triggered by peaks of [Ca2+]i of ∼10 μmol/l at the release sites (16,26). In a new study, the effects of several exogenous Ca2+ chelators on insulin release were analyzed to know how cytosolic Ca2+ distribution affected the dynamics of insulin exocytosis (27). These experiments were also supported by modeling Ca2+ diffusion in the vicinity of channels and release sites in conditions that simulated trains of depolarizations that resembled those induced by stimulatory glucose concentrations. The findings obtained in this study suggested that the first phase of glucose-induced insulin release is due to exocytosis of a primed pool of secretory granules located in close proximity to Ca2+ channels (around 50 nm), whereas the second phase is due to mobilization of a reserve pool of granules placed at ∼300 nm from the plasma membrane. In addition, this model predicted that [Ca2+]i in the outermost shells of pancreatic β-cells was in the micromolar range but not higher than 14 μmol/l. These results were consistent with other reports (16,19,26) and provided additional evidence in favor of the existence of two different vesicle types responsible for glucose-induced insulin release: primed vesicles, located close to the Ca2+ channels, and reserve vesicles, not strictly co-localized with the Ca2+ channels.
Later on, a new report using a Monte Carlo simulation of three-dimensional buffered Ca2+ diffusion (28,29) demonstrated again that a large free Ca2+ concentration was localized near the ion channels, with steeper gradients in the submembrane domain. Theoretical predictions were then further supported by experimental data obtained using spot confocal microscopy, a technique that excels in measuring spatial-restricted intracellular Ca2+ signals because of its remarkable high signal-to-noise ratio (15,30). This technique enabled us to measure and characterize the glucose-induced [Ca2+]i gradients in the first two microns close to the plasma membrane to verify the values simulated by models (15). These results demonstrated that, in pancreatic islet cells, nutrient secretagogues induce Ca2+ microgradients, which dissipate within the first micron from the membrane, and that [Ca2+]i can readily reach around 10 μmol/l (Fig. 2). The finding that these gradients were maintained during the period that the stimulus was present also indicated that pancreatic islet cells might have a specialized and compartmentalized Ca2+ homeostatic machinery, mainly controlled by the mitochondrias and the endoplasmic reticulum (Fig. 1). These intracellular organelles are likely to be in close proximity to the plasma membrane, shaping these Ca2+ gradients and restricting the action of the Ca2+ signal to the exocytotic site (15).
cGMP: A NEW PLAYER IN THE REGULATION OF KATP ACTIVITY
The endocrine pancreas is not considered to be a classic estrogen target, although the effects of 17β-estradiol on some physiological aspects of the islets of Langerhans have been known for a long time. On the one hand, the level of plasma insulin is increased in pregnant rats in response to increased levels of sex steroids (31–33). Moreover, 17β-estradiol at concentrations comparable to those of pregnancy enhances insulin secretion in perfused rat pancreas (34). In humans, 17β-estradiol reverses the effect of menopause on glucose and insulin metabolism, resulting in an increase of pancreatic insulin secretion as well as an improvement of insulin resistance (35,36). On the other hand, in glucagon-releasing α-cells, it was described that 17β-estradiol produces an inhibitory effect on glucagon secretion (37). In spite of this evidence, the mechanism of action used by 17β-estradiol on α- and β-cells is just being discovered.
Recently, an estrogen membrane receptor was visualized in pancreatic β-cells. It is responsible for a rapid insulinotropic effect of 17β-estradiol when applied at physiological concentrations (38). Once bound to its membrane receptor, estrogen triggers the synthesis of cGMP, which in turn activates protein kinase G (PKG). Then, the KATP channels close in a PKG-dependent manner, causing the plasma membrane to depolarize, enhancing intracellular calcium signals (5). As a result, insulin secretion is increased (38). This receptor not only exists in pancreatic β-cells, but also in α-cells. When 17β-estradiol acts through this receptor in α-cells, it inhibits low glucose-induced [Ca2+]i oscillations, and therefore it abolishes glucagon release (39).
The cGMP-PKG pathway is not exclusive of the membrane effect of 17β-estradiol, because it is also used by glucose to induce insulin secretion. It has been reported that glucose induces a dose-response increase on cGMP levels in islets of Langerhans (40). Furthermore, inhibitors of cGMP synthesis block both the insulin secretion (41,42) and the intracellular calcium oscillatory pattern (Fig. 3) induced by glucose. Although it is not known whether the increase in cGMP levels after glucose metabolism directly affects the KATP activity, estradiol certainly produces a decrease in its activity through a PKG-dependent mechanism (5,38) (Fig. 4.). This points to the potassium channel as the target for the cGMP-PKG pathway activated by glucose and estradiol, which in turn produces the depolarization of the plasma membrane, the increase in the intracellular calcium, and the insulin release. However, whether the KATP is phosphorylated by PKG or is closed by an indirect mechanism is still unknown. The fact that the estradiol action is mediated by the same intracellular pathway as the glucose highlights the importance of the hormone as a potentiator of the insulinotropic effect of the nutrient.
NONCLASSICAL MEMBRANE ESTROGEN RECEPTORS IN ISLET CELLS
The nature of the membrane estrogen receptor responsible for the rapid effects known to be produced by estradiol in a large number of cell types has been a matter of controversy in recent years. Whereas some groups support the view that the intracellular estrogen receptor (ER)-α can be located at the plasma membrane, it still remains unclear how this soluble intracellular receptor can be attached to the membrane (43). However, pharmacological studies of the islets of Langerhans demonstrate that the membrane ER responsible for the physiological effects of 17β-estradiol is not related to any of the intracellular ERs described so far. This was shown by the use of several antibodies against ER-α and ER-β, which failed to recognize any structure on the plasma membrane of the cells (44). Instead, the membrane ER seems to be an undefined receptor that binds adrenaline, norepinephrine, and dopamine (39) but does not interact with either specific anti-estrogens such as ICI182,780 (39) or other steroids such as estriol (44). This nonclassical membrane estrogen receptor is not related to adrenergic or dopaminergic receptors, as demonstrated by the lack of interaction with specific ligands for these receptors. Many chemicals were tested in binding experiments, and a structural feature was obtained from those able to block the interaction between 17β-estradiol and nonclassical membrane ER in nonpermeabilized cells. Surprisingly, it was found that the nonclassical membrane ER binds molecules with either a catechol ring or the A-ring of the 17β-estradiol within its structure, such as 2-hydroxyestradiol (39,44).
NUCLEAR Ca2+ SIGNALS AND GENE EXPRESSION
In many different types of cells, including the pancreatic β-cell, Ca2+ changes in the nucleus are believed to be an important regulator of gene expression (45,46). Because nuclear processes exhibit a significant Ca2+-dependent responsiveness, the regulation of Ca2+ within the nucleus should be strictly controlled. However, the mechanisms whereby nuclear Ca2+ signals are induced and regulated are still controversial. Whereas it has been shown that functional channels in the nuclear membrane can regulate nuclear Ca2+ (46,47), some groups have argued against an independent nuclear Ca2+ homeostasis because of the presence of nuclear pores, which would allow for a rapid Ca2+ equilibration between nucleus and cytoplasm (47).
In pancreatic β-cells, besides the well-known effect on insulin release, Ca2+ influx by membrane depolarization induced by nutrient metabolism can also activate gene transcription (48). In fact, high glucose and fatty acids can regulate the expression of immediate-early response genes such as c-fos, c-jun, junB, zif-268, and nur-77, which are also coding for transcription factors implicated in several functions such as cell proliferation and differentiation (49,50). It has also been shown that long exposure of β-cells to high glucose levels increase the expression of genes coding for some metabolic enzymes (51). The expression of most of these genes, whose induction is mediated by nutrients, has in common the participation of a Ca2+ signal (52). However, the mechanisms that link these extracellular messages with nuclear function have been explored little. Recently, a novel pathway was reported by which nutrients could transmit Ca2+ messages to the nucleus that activate nuclear function (46). In this study, it was shown that a KATP channel with similar properties to that found on the plasma membrane is also present on the nuclear envelope of pancreatic β-cells. Blockade of nuclear KATP channels with the sulfonylurea tolbutamide or with DPs triggered nuclear Ca2+ transients by a voltage-sensitive mechanism (possibly ryanodine receptors), which induced phosphorylation of the transcription factor CREB (cAMP response element binding protein) (Fig. 4). In whole cells, fluorescence in situ hybridization also revealed that these Ca2+ signals might trigger c-myc expression. Thus, in the pancreatic β-cell, the increase of the ATP/ADP ratio, DPs, and possibly cGMP, as a result of nutrient metabolism, may act not just on plasma membrane KATP channels inducing extracellular Ca2+ influx, but also may affect those channels located in the nucleus producing nuclear Ca2+ transients. These novel findings indicate that functional KATP channels in the nucleus could be critical to link nutrient metabolism and nuclear function by Ca2+ signals. In view of the fact that, in several systems, gene expression is modulated by the code established by the amplitude, duration, and/or frequency of Ca2+ signals (53), it would be particularly interesting to analyze in future studies the relation between the dynamics of Ca2+ in the nucleus and its effect on nuclear function in the pancreatic β-cell. Actually, preliminary results of our group indicate that variations in the amplitude and duration of Ca2+ nuclear levels could play a role in the regulation of the expression of genes such as c-fos and c-myc.

Model for stimulus-secretion coupling in the pancreatic β-cell. The classic stimulus-secretion coupling pathway that mediates insulin release involves the closure of plasma membrane KATP channels as a result of glucose (G) metabolism by increasing both the intracellular ATP/ADP ratio and DPs. Channel closure leads to membrane depolarization (↑Vm), which activates voltage-operated Ca2+ channels, producing a corresponding Ca2+ influx. This cytosolic Ca2+ signal mediates granule exocytosis and insulin release. The regulation of the Ca2+ signal is further achieved by the joint action of plasma membrane Ca2+ pumps and, particularly, subcellular organelles such as mitochondrias and the endoplasmic reticulum, which participate in shaping and confining the Ca2+ signal by uptake/release mechanisms.
Model for stimulus-secretion coupling in the pancreatic β-cell. The classic stimulus-secretion coupling pathway that mediates insulin release involves the closure of plasma membrane KATP channels as a result of glucose (G) metabolism by increasing both the intracellular ATP/ADP ratio and DPs. Channel closure leads to membrane depolarization (↑Vm), which activates voltage-operated Ca2+ channels, producing a corresponding Ca2+ influx. This cytosolic Ca2+ signal mediates granule exocytosis and insulin release. The regulation of the Ca2+ signal is further achieved by the joint action of plasma membrane Ca2+ pumps and, particularly, subcellular organelles such as mitochondrias and the endoplasmic reticulum, which participate in shaping and confining the Ca2+ signal by uptake/release mechanisms.
![FIG. 2. [Ca2+]i changes beneath the plasma membrane. In the scheme, we illustrate the existence of submembranal [Ca2+]i gradients elicited by glucose that can reach Ca2+ levels in the micromolar range in the first micron beneath the plasma membrane. [Ca2+]i decays to hundreds of nanomoles in the bulk cytosol. The figure was elaborated based on the data published by Quesada et al. (15).](https://ada.silverchair-cdn.com/ada/content_public/journal/diabetes/53/suppl_1/10.2337_diabetes.53.2007.s86/2/m_db02t0013002.jpeg?Expires=1716520334&Signature=sQfG19HqD1TsbpWKjX85lR-QHSLYtSGxxrAtyQS~xUT~SqTY5UdijUMA5sWZ7H4w3RgwzQycfmvJDLVWig4TJll5BRlxwPvvFHXZO~dIH1mFDpfJS4cVq7Py02zctVDNDtLVwZbLZj5FAXpUnJXsusJ6C21A4xw6Ez9Kobuv5qEMVrKQFneKAwON9yH4GGESNCI3H4CuVUexH2stHffZVxhTs2pZICcNsZT4XoLYFZ4lCKBPHke4Hiya1Ff61XyOPGlCyt7CdxFhUOcwpiHALIyoXwM-MK981FFVeIv70ELa8ya-FLTcu8EKo9zUmmTNSPe4d-L8N4RaCQoeseipsQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
[Ca2+]i changes beneath the plasma membrane. In the scheme, we illustrate the existence of submembranal [Ca2+]i gradients elicited by glucose that can reach Ca2+ levels in the micromolar range in the first micron beneath the plasma membrane. [Ca2+]i decays to hundreds of nanomoles in the bulk cytosol. The figure was elaborated based on the data published by Quesada et al. (15).
[Ca2+]i changes beneath the plasma membrane. In the scheme, we illustrate the existence of submembranal [Ca2+]i gradients elicited by glucose that can reach Ca2+ levels in the micromolar range in the first micron beneath the plasma membrane. [Ca2+]i decays to hundreds of nanomoles in the bulk cytosol. The figure was elaborated based on the data published by Quesada et al. (15).

Oscillatory intracellular calcium pattern in response to 11 mmol/l glucose in a mouse islet of Langerhans. A total of 10 μmol/l of LY83,583, an inhibitor of the soluble guanylate cyclase, blocks the oscillations.
Oscillatory intracellular calcium pattern in response to 11 mmol/l glucose in a mouse islet of Langerhans. A total of 10 μmol/l of LY83,583, an inhibitor of the soluble guanylate cyclase, blocks the oscillations.
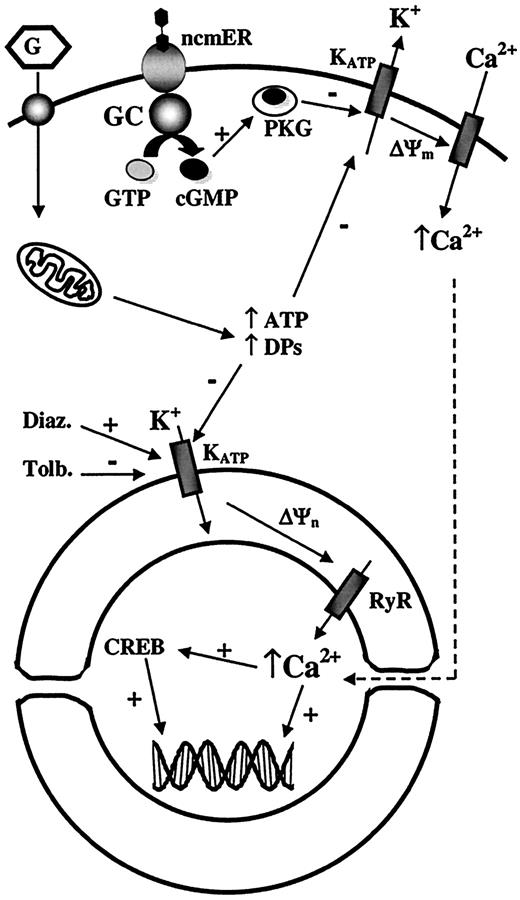
Proposed model for the role of KATP channels on the nuclear membrane. ATP/ADP ratio and DPs are increased as a consequence of glucose metabolism. This increase closes plasma membrane KATP channels producing the depolarization of the plasma membrane, but also affects those KATP channels located on the nuclear membrane. Drugs such as tolbutamide and diazoxide can also act on nuclear KATP channels. Blockade of these channels produces a rise in nuclear transmembrane potential (ΔΨn), which leads to a Ca2+ release from the nuclear envelope to the nucleoplasm mainly through ryanodine receptor Ca2+ release channels (RyR). These nuclear Ca2+ signals modulate nuclear functions such as CREB (cAMP response element binding protein) phosphorylation and likely gene expression. The effect of 17β-estradiol upon binding to the nonclassical membrane estrogen receptor (ncmER) is also illustrated. The activation of this receptor enhances the production of cGMP, which in turn activates PKG. Subsequently, KATP channels close in a PKG-dependent manner, producing membrane depolarization and enhancing Ca2+ influx. ΔΨm, plasma membrane potential; Diaz., diazoxide; GC, guanylate cyclase; Tolb., tolbutamide.
Proposed model for the role of KATP channels on the nuclear membrane. ATP/ADP ratio and DPs are increased as a consequence of glucose metabolism. This increase closes plasma membrane KATP channels producing the depolarization of the plasma membrane, but also affects those KATP channels located on the nuclear membrane. Drugs such as tolbutamide and diazoxide can also act on nuclear KATP channels. Blockade of these channels produces a rise in nuclear transmembrane potential (ΔΨn), which leads to a Ca2+ release from the nuclear envelope to the nucleoplasm mainly through ryanodine receptor Ca2+ release channels (RyR). These nuclear Ca2+ signals modulate nuclear functions such as CREB (cAMP response element binding protein) phosphorylation and likely gene expression. The effect of 17β-estradiol upon binding to the nonclassical membrane estrogen receptor (ncmER) is also illustrated. The activation of this receptor enhances the production of cGMP, which in turn activates PKG. Subsequently, KATP channels close in a PKG-dependent manner, producing membrane depolarization and enhancing Ca2+ influx. ΔΨm, plasma membrane potential; Diaz., diazoxide; GC, guanylate cyclase; Tolb., tolbutamide.
This article is based on a presentation at a symposium. The symposium and the publication of this article were made possible by an unrestricted educational grant from Les Laboratoires Servier.
Article Information
This study was partially supported by grants from Ministerio de Ciencia y Tecnología, the Juvenile Diabetes Research Foundation International, Generalitat Valenciana, Fundació Marató TV3, the European Foundation for the Study of Diabetes, and Instituto Carlos III (RET:p G03/171, RGDM G03/212, TERCEL G03/210, RCMN C03/08).